Publié le 28 sep 2010Lecture 5 min
Maladie de Sandhoff (gangliosidose GM2)
R. ABILKASSEM, A. AGADR, N. DINI, H. EN-NOUALI Hôpital Militaire, Hôpital d’Enfants Rabat
La maladie de Sandhoff est une maladie neurométabolique rare, de transmission autosomique récessive, due au déficit en deux hydrolases lysosomale ; la β-hexosaminidase A et la β-hexosaminidase B(1,2). La forme infantile est la plus sévère. Nous rapportons le cas d’un nourrisson de 18 mois né de parents consanguins et ayant un frère décédé à l’âge de 16 mois.
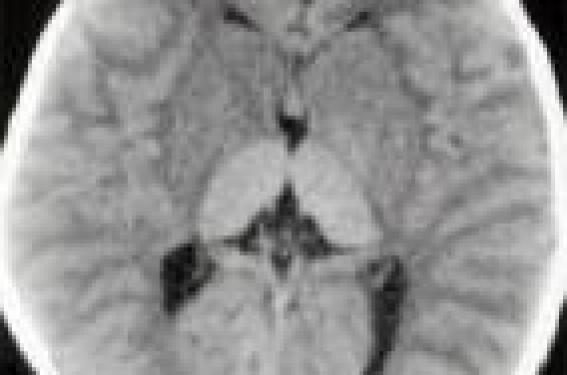
Observation D… âgé de 18 mois est hospitalisé pour exploration d’une régression psychomotrice et une macrocéphalie. Il est issu de parents consanguins de premier degré. Dans ses antécédents on trouve un frère décédé à l’âge de 16 mois d’une pathologie neurologique semblable. D… est né à terme avec une bonne adaptation à la vie extra-utérine, un poids de 3,3 kg et un périmètre crânien de 36 cm. Durant les 5 premiers mois de la vie, le développement psychomoteur était satisfaisant pour l’âge avec une tenue de la tête à 3 mois, une bonne poursuite oculaire, il sourit avec les parents. Par la suite, il a présenté un syndrome fébrile non expliqué, une régression psychomotrice avec perte de la tenue de la tête, suivi d’une hypotonie axiale, une perte du sourire réponse et de la poursuite oculaire. Les parents ont constaté également des sursauts audiogènes et une augmentation progressive du périmètre crânien. L’examen trouve une macrocéphalie avec un périmètre crânien à 51 cm (+2 DS). Il n’y a pas de syndrome dysmorphique, pas de poursuite oculaire ni de réactivité à la lumière. On observe une hypotonie axiale, un syndrome pyramidal avec un signe de Babinski bilatéral, des accès paroxystiques de type myoclonique permanents intéressant l’hémiface droit avec l’hémicorps homolatéral, suivis d’une généralisation a tout le corps. Il n’y a pas d’hépatosplénomégalie. Le fond d’oeil objective une tache rouge cerise maculaire bilatérale (figure 1/illustration). Le scanner cérébral montre une hyperdensité bilatérale des thalamus avec une hypodensité accentuée de la substance blanche (figure 2). Figure 2. TDM cérébrale : hyperdensité bilatérale du thalamus avec une hypodensité accentuée de la substance blanche. L’imagerie par résonance magnétique cérébrale (IRM) retrouve un hyposignal au niveau du thalamus en T1 et un hypersignal en T2 (figure 3). Le tracé électroencéphalographique (EEG) montre un tracé ralenti sans pattern EEG spécifique. Le dosage des hexosaminidases A et B dans le sérum et les leucocytes témoigne d’une activité nulle, confirmant le diagnostic de maladie de Sandhoff. Discussion La maladie de Sandhoff est une maladie de surcharge lysosomale, qui appartient à la famille des gangliosidoses à GM2. Elle est liée à l’absence de la dégradation des gangliosides GM2 par les hexosaminidases A et B et à leur accumulation essentiellement dans les neurones et les tissus périphériques (1,3). La maladie de Sandhoff est une maladie neurométabolique rare, de transmission autosomique récessive, bien que des cas sporadiques aient été rapportés. Sa fréquence en Europe est estimée à 1/130 000 naissances vivantes. Le gène responsable de la mutation est localisé sur le chromosome 5 (5q13). Les hexosaminidases A et B sont formées par la dimérisation de deux sous-unités α et β. La β-hexosaminidase A contient les sous-unités α et β, alors que la β-hexosaminidase B se compose de deux sous-unités β. Le déficit en sousunité α est responsable de la maladie de Tay Sach, alors que le déficit, voire l’absence de la sousunité β est responsable de la maladie de Sandhoff désignée aussi sous le nom de la variante O. Trois tableaux cliniques variés sont décrite (infantile, juvénile, adulte) selon l’âge d’apparition des symptômes ainsi que la sévérité des signes cliniques ou du pronostic (3). Figure 3. IRM cérébrale : hyposignal au niveau du thalamus en T1 et hypersignal en T2. La forme infantile débute entre 3 et 6 mois de vie par une régression des acquisitions psychomotrices, une hypotonie, des clonies audiogènes, des taches rouge cerise au niveau de la macula. L’évolution est marquée par une dégradation neurologique rapide avec apparition d’une tétraparésie spastique, des crises épileptiques de types variés, une macrocéphalie progressive et une cécité. L’atteinte du système nerveux central se manifeste radiologiquement par une hyperdensité bilatérale du thalamus, avec une hypodensité accentuée de la substance blanche sur le scanner cérébral, et un hyposignal des thalamus avec un hypersignal au niveau de la substance blanche cérébrale sur la séquence pondéré T2 à l’IRM cérébrale (4-6). Le diagnostic est confirmé par le dosage des hexosaminidases A et B dans le sérum, les leucocytes et les fibroblastes, qui montre une activité nulle (7). Pronostic Il n’existe pas de traitement curatif radical, le traitement reste symptomatique, le pronostic est sombre, l’espérance de vie dépassant rarement 4 ans (1,3). Le diagnostic prénatal reste possible dès la 8e semaine de gestation par la mesure de l’activité enzy-matique au niveau des cellules villositaires. La recherche de mutations géniques peut fournir des informations valables pour une consultation prénuptiale surtout dans les groupes à risque. C’est souvent à la suite de cas familiaux qu’on peut entreprendre cette démarche.
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :