Publié le 08 mar 2010Lecture 16 min
Maladie de Rendu-Osler : diagnostic et conduite à tenir
H. ADAMSKI*/***, R. CORRE**/*** *Service de dermatologie **Service de pneumologie ***Centre de compétence de la maladie de Rendu-Osler CHU Pontchaillou, Rennes
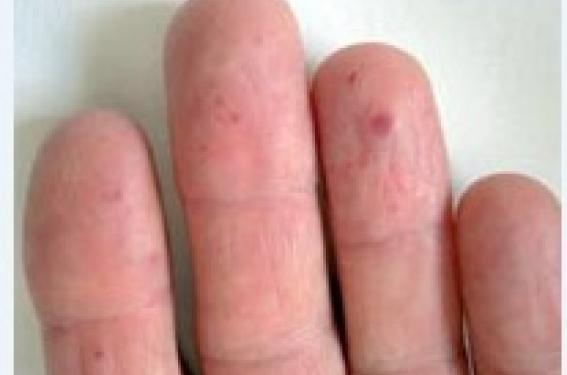
La télangiectasie hémorragique héréditaire, encore appelée maladie de Rendu-Osler (MRO) du nom de ses premiers descripteurs, est une maladie rare. Sa prévalence est estimée à 1/10 000. Le diagnostic de MRO est clinique et repose sur l’association : télangiectasies cutanées, épistaxis et manifestations viscérales et hérédité. Il s’agit d’une maladie génétique à transmission autosomique dominante d’expressivité variable bien souvent révélée par ses complications viscérales.
Génétique et physiopathologie (1) Dans la MRO, la transmission autosomique dominante concerne plus de 90 % des patients, les autres cas correspondant à des mutations de novo. L’expressivité est variable (les manifestations cliniques varient d’un individu à l’autre). La pénétrance est liée à l’âge et elle est complète entre 40 et 50 ans : ainsi les signes cliniques, qui sont habituellement absents à la naissance, apparaissent progressivement avec l’âge. Les épistaxis sont le premier symptôme pouvant apparaître dès l’enfance puis les malformations artérioveineuses (MAV) pulmonaires à l’adolescence et ensuite les télangiectasies cutanées ou viscérales à l’âge adulte. Les épistaxis sont le premier symptôme pouvant exprimer la maladie dès l’enfance. L’étude génétique a mis en évidence au moins deux gènes impliqués dans la maladie qui codent des protéines interagissant avec la superfamille TGF (transforming growth factor)-β : facteur déterminant de l’intégrité de la paroi vasculaire. Le premier localisé sur le chromosome 9 (9q33-q34) ENG code pour l’endogline et le deuxième sur le chromosome 12 (19q11-14) ACVLR1 pour un récepteur membranaire de l’activine à activité kinase ALK-1. Plus récemment, des mutations ont été décrites au niveau d’un troisième gène (MADH4 en position 18q21.1) codant pour la protéine SMAD4 chez les patients présentant une polypose juvénile familiale et pour certains une MRO. L’endogline et ALK-1 sont des récepteurs membranaires pour le TGF-β exprimé à la surface des cellules endothéliales. La protéine SMAD4 participe à la signalisation intracellulaire par la voie du TGF-β. Le développement de vaisseaux dystrophiques au cours de la MRO serait consécutif aux anomalies de signalisation intracellulaire par la voie du TGF-β. La participation de plusieurs gènes expliquerait en partie l’hétérogénéité de la maladie. Les mutations du gène ENG seraient responsables d’un risque plus élevé d’épistaxis plus sévères et précoces ainsi que du risque de développer une MAV pulmonaire. Les MAV hépatiques seraient plus fréquentes en cas de mutation ACVLR1. Diagnostic Actuellement, le diagnostic de MRO reste clinique et morphologique et ses critères ont été redéfinis selon la conférence de consensus de Curaçao (tableau 1). Il se fonde sur l’association : télangiectasies cutanées, épistaxis spontanées et récurrentes, de localisations viscérales et d’antécédents familiaux de MRO : le diagnostic est considéré comme formel lorsque 3 au moins des critères sont présents (2). Les manifestations viscérales présentent la particularité d’être souvent asymptomatiques mais peuvent donner lieu à des complications graves. Le médecin et le patient doivent en être informés afin de proposer des mesures de diagnostic et de prise en charge adéquates. Manifestations cutanéomuqueuses (1,3) Épistaxis Les épistaxis sont dues à un saignement spontané de télangiectasies de la muqueuse nasale. Elles représentent la manifestation la plus commune de la MRO retrouvée chez plus de 90 % des patients. Les épistaxis récurrentes débutent vers l’âge de 10 ans chez de nombreux patients. Elles sont classiquement récidivantes, parfois sévères et abondantes. Ces épistaxis peuvent entraîner un syndrome anémique et nécessiter de multiples transfusions et une supplémentation orale ou intraveineuse en fer. Télangiectasies cutanéomuqueuses Les télangiectasies se présentent sous l’aspect de macules rougeâtres, pourpres ou violacées, bien limitées, arrondies en tête d’épingle, pâlissant à la vitro-pression. Les lésions prennent un aspect parfois en relief avec le temps : « point rubis ». Figure 2. Maladie de Rendu-Osler : télangiectasies papuleuses du menton, de la joue gauche et de la lèvre inférieure. Figure 1. Maladie de Rendu-Osler : télangiectasies de la joue droite. Des angiomes stellaires, de simples dilatations capillaires ou veineuses peuvent également être observés. Les télangiectasies cutanées apparaissent typiquement plus tardivement que les épistaxis, classiquement entre 20 et 30 ans, et se multiplient souvent par poussées, notamment pendant la grossesse. Elles sont présentes chez les trois quarts des patients atteints de MRO. Les lésions cutanées siègent électivement sur l’extrémité céphalique (figures 1 et 2) : joues, nez, lèvres, oreilles, front, menton, paupières. Le cuir chevelu est généralement respecté. Des lésions peuvent s’observer aux membres notamment au niveau des mains (figure 3). Il peut exister des saignements de ces télangiectasies cutanées, mais ils sont rarement importants. Les télangiectasies cutanées apparaissent classiquement entre 20 et 30 ans. Les lésions muqueuses concernent plus fréquemment les fosses nasales, la paroi pharyngée et la cavité buccale (figure 4) : bord et dos de la langue, face interne des joues, gencives, palais. Les télangiectasies correspondent au début à des dilatations des veinules postcapillaires dermiques puis deviennent des communications artériolo- veinulaires avec disparition des segments capillaires. Manifestations viscérales Manifestations pulmonaires (3,4) Figure 3. Maladie de Rendu-Osler : télangiectasies digitales. Elles sont présentes dans 30 % des cas et sont dues essentiellement aux MAV pulmonaires. Ces malformations, souvent multiples, prédominent dans les lobes inférieurs et sont de taille très variable. Elles consistent en des communications directes d’une branche de l’artère et de la veine pulmonaire à travers un anévrysme à parois fines entraînant un shunt droitegauche. Les complications sont de deux types : • Fonctionnelles : - avec création d’un shunt droitgauche pouvant entraîner une hypoxémie à l’origine d’une dyspnée d’effort qui est le symptôme respiratoire le plus fréquent. Parfois, elle s’exprime lors du passage de la position horizontale à la position verticale : on parle de platypnée. Figure 4. Maladie de Rendu-Osler : multiples télangiectasies de la langue. Elle s’explique par une localisation préférentielle des MAV aux bases pulmonaires, la position debout entraîne une augmentation de l’afflux sanguin dans les bases, ce qui aggrave l’effet shunt. L’hypoxémie peut également entraîner une cyanose et l’apparition d’un hippocratisme digital. Un souffle auscultatoire peut être entendu en regard de la MAV ; - la perte du « filtre » capillaire peut être responsable d’embolie cruorique ou septique. Des microemboles échappent, à travers la MAV pulmonaire au filtre capillaire, et atteignent ainsi les artères systémiques, le plus souvent le cerveau, à l’origine d’abcès cérébraux, d’accidents ischémiques transitoires ou constitués. Environ 25 à 30 % des patients porteurs de MAV pulmonaires dans le cadre d’une MRO présenteront une symptomatologie cérébrale ischémique. • Mécaniques : les vaisseaux anormaux peuvent se rompre spontanément dans 9 à 13 % des cas dans une bronche ou dans une cavité pleurale, entraînant une hémoptysie ou un hémothorax d’abondance variable. La rupture spontanée des vaisseaux serait favorisée par la grossesse du fait de l’augmentation de la volémie et du débit cardiaque. L’intérêt du dépistage des MAV pulmonaires est donc fondamental en cas de MRO définie ou possible. La tomodensitométrie thoracique spiralée est l’examen de référence pour affirmer la présence de MAV (figure 5). Figure 5. Maladie de Rendu-Osler : malformation artérioveineuse du lobe inférieur pulmonaire gauche mesurant 3 cm avec présence de vaisseaux afférents et efférents, visualisée en tomodensitométrie (flèche) qui a été ensuite embolisée. Afin de mettre en évidence un effet shunt droitegauche, plusieurs examens sont réalisés. La mesure de la PaO2 et de SaO2 par gazométrie artérielle : une hypoxémie témoigne de l’existence de MAV pulmonaire, mais elle peut être normale si le shunt est modéré. Le facteur de transfert du CO peut être abaissé. Le dépistage des MAV pulmonaires est fondamental en cas de MRO définie ou possible. L’échographie cardiaque de contraste est un des examens clés actuels, très sensibles et non invasifs pour la recherche du shunt non détecté par la gazométrie. En présence de MAV pulmonaire, les bulles ne sont plus arrêtées par le capillaire pulmonaire et apparaissent dans l’oreillette gauche 3 à 8 cycles cardiaques (2 à 5 secondes) après avoir quitté le ventricule droit. En cas de shunt intracardiaque, les bulles apparaissent immédiatement dans l’oreillette gauche. Les MAV pulmonaires tendent à augmenter de taille, surtout si elles sont multiples et en cas de grossesse et régressent rarement spontanément. L’hypertension artérielle pulmonaire est rare (< 1 %) au cours de MRO. Elle peut résulter d’une augmentation du débit cardiaque du fait des MAV systémiques. Manifestations neurologiques Les manifestations neurologiques sont observées dans 8 à 12 % des cas. Elles peuvent être observées à tout âge, avec un pic d’incidence dans les 3e et 4e décennies. Elles résultent de deux mécanismes physiopathologiques distincts : – les MAV pulmonaires, déjà évoquées plus haut, font perdre au poumon sa fonction de « filtre », favorisant ainsi le passage d’emboles dans la circulation cérébrale, responsable des deux tiers des manifestations neurologiques observées dans la MRO (abcès cérébraux, méningites purulentes, accidents ischémiques transitoires ou constitués) ; – les malformations vasculaires cérébro-médullaires : il peut s’agir de malformations caverneuses, d’angiomes veineux, de MAV. Leur incidence n’est pas clairement définie. Elles peuvent être, après rupture, à l’origine d’hémorragies sous-arachnoïdiennes ou intracérébrales (5). Manifestations digestives et hépatiques L’incidence des manifestations digestives est estimée entre 11 et 40 % des cas. Elles sont tardives, et surviennent après 50 ans. Il s’agit d’hémorragies récurrentes du tractus gastro-intestinal se produisant sous la forme de mélaena le plus souvent et se compliquant d’anémie. Par ordre de fréquence, elles sont dues à des télangiectasies de l’estomac, du duodénum, de l’oesophage et enfin de l’intestin. L’aspect endoscopique est la télangiectasie capillaire sous-muqueuse, ronde et rouge assez proche de l’aspect des lésions cutanées (6). L’incidence des manifestations hépatiques est estimée à 8-16 % des cas. C’est une manifestation tardive (après 60 ans). Les signes cliniques sont le plus souvent très discrets : hépatomégalie sensible et souffle vasculaire dans l’hypochondre droit. Les troubles sont corrélés à l’importance et la nature des MAV hépatiques (sus-hépatique, porte) et à l’origine d’insuffisance cardiaque par hyperdébit, d’hypertension portale ou d’encéphalopathie hépatique. Les facteurs aggravants sont l’anémie, la fibrillation auriculaire et le dernier trimestre de la grossesse. En dehors de ces épisodes aigus, la biologie hépatique ne montre qu’une élévation modérée des phosphatases alcalines et des γGT (7). Diagnostic différentiel Le diagnostic différentiel de MRO est multiple et se fait en fonction de symptômes participant au diagnostic, comme les télangiectasies, les épistaxis ou les malformations artérioveineuses viscérales. Dans ce paragraphe, seront abordées les principales affections caractérisées par des télangiectasies. On distingue les formes acquises et héréditaires (8). Télangiectasies acquises La rosacée, notamment dans sa phase érythro-couperosique (figure 6). Les télangiectasies en forme d’étoile vasculaire ou angiome stellaire (centré par un point rouge dont s’échappent des arborisations centrifuges). Elles sont accompagnées d’érythème et parfois de pustules qui surviennent brusquement par poussées après certains facteurs déclenchants (repas chauds, prise d’alcool, stress, exposition solaire, changements brutaux de température, etc.). Figure 6. Angiomes stellaires des joues au cours d’une rosacée. Les lésions siègent sur le visage, particulièrement au niveau du nez et des joues, sans atteinte de la muqueuse buccale. Le CREST syndrome est une forme de sclérodermie avec calcinoses sous-cutanées digitales, syndrome de Raynaud et atteinte oesophagienne. Les télangiectasies siègent au niveau du visage (figure 7) et des doigts et elles sont très proches cliniquement de la MRO (9). Cette affection survient souvent après 40 ans. Il n’y a pas d’épistaxis associée. La grossesse et les cirrhoses hépatiques (figure 8) sont des situations associées à l’apparition de télangiectasies, habituellement de type angiome stellaire : le rôle de facteurs hormonaux est postulé dans les deux cas. Figure 7. Télangiectasies au cours d’une sclérodermie. Citons la forme télangiectasique des mastocytoses, qui est souvent méconnue car la turgescence par frottement peut manquer. Télangiectasies héréditaires Les télangiectasies héréditaires bénignes se distinguent difficilement des télangiectasies de la MRO. Elles sont souvent précoces dans l’enfance, d’évolution lente et ont un aspect plutôt en étoile. Figure 8. Érythème avec étoiles vasculaires multiples du décolleté au cours d’une cirrhose (angiomes stellaires). C’est surtout l’absence d’atteinte des muqueuses et systémique associée qui oriente vers ce diagnostic. Les télangiectasies héréditaires bénignes se distinguent difficilement des télangiectasies de la MRO. ● L’ataxie-télangiectasie est une maladie autosomique récessive rare comportant une ataxie cérébelleuse, un déficit immunitaire et des télangiectasies de type chevelu capillaire survenant vers 3 à 5 ans au niveau de la conjonctive, du nez, des oreilles et au pli des coudes. Les autres signes sont : taches café au lait, eczéma, photosensibilité et cancers cutanés et viscéraux. Le gène muté dans cette maladie code une enzyme qui phosphoryle la protéine p53. Stratégie de dépistage et de surveillance • Chez les patients atteints de MRO, une prise en charge diagnostique des membres de la famille est à réaliser. Les recherches mutationnelles des gènes ALK1 et ENG sont à proposer à tout patient dont le diagnostic est cliniquement certain ou possible et aux sujets apparentés à un sujet atteint chez qui la mutation familiale responsable de la MRO a été identifiée. • La recherche de MAV pulmonaire est systématique chez les malades et les membres de leur famille en raison des complications pulmonaires et neurologiques par radiographie thoracique, tomodensitométrie spiralée et échographie cardiaque de contraste. Chez un patient asymptomatique, les examens de dépistage doivent être répétés, pour s’assurer de l’absence de nouvelles MAV (3). • Le dépistage des malformations vasculaires neurologiques doit comporter tout d’abord un interrogatoire et un examen clinique orienté(5). La réalisation d’une angio-IRM cérébrale et spinale pour le dépistage de MAV est recommandée chez les femmes en âge de procréer avant toute grossesse et peut être proposée chez les nourrissons atteints de la maladie. • Pour la recherche de MAV hépatique, il sera effectué un bilan biologique (γGT, phosphatases alcalines, transaminases) afin d’évaluer le retentissement hépatique et la confirmation de l’absence du virus de l’hépatite B (patients à haut risque transfusionnel). Les examens d’imagerie comporteront une échographie et un Doppler hépatique avec mesures du diamètre des vaisseaux et des vitesses de flux et/ou un scanner ou une IRM. Une échographie cardiaque de départ sera réalisée pour en évaluer le retentissement des MAV hépatiques sur la fonction myocardique (7). • Il n’y a pas de bénéfice à dépister systématiquement des angiomes digestifs sans signe d’appel, car les actions préventives n’ont pas montré actuellement leur utilité (6). • En raison du risque de poussées évolutives de la MRO lors de certaines périodes de la vie du patient, la surveillance doit être renforcée pendant la grossesse qui majore le risque de MAV. Traitement ● Épistaxis La grande majorité des épistaxis en phase aiguë est contrôlée par tamponnement antérieur et postérieur. Le traitement préventif fait appel à différentes techniques telles que les cautérisations chimiques ou électriques, la cryothérapie à l’azote liquide, le laser CO2, Yag ou KTP, les injections in situ d’éthibloc, les injections sousmuqueuses de colle biologique, la dermatoplastie chirurgicale endonasale, les ligatures artérielles, l’embolisation des deux pédicules sphénopalatins et des deux artères faciales, la fermeture bilatérale des fosses nasales (10). ● Télangiectasies cutanées Les traitements ont longtemps été limités à l’électrocoagulation et à la cryothérapie avec risque d’un résultat imparfait et de séquelles cicatricielles. Ils sont dominés actuellement par le développement des techniques lasers « vasculaires » (KTP, colorant pulsé, etc.), progrès indiscutable, qui donnent de bons résultats(8). Les mesures préventives qui visent à diminuer l’intensité et la fréquence des récidives sont à proposer aux patients (photoprotection, arrêt d’une corticothérapie injustifié, etc.). ● Manifestations pulmonaires Chez les sujets porteurs de MAV pulmonaires, une antibioprophylaxie (β-lactamines ou synergistines en cas d’allergie aux pénicillines) est recommandée en cas de gestes invasifs (intervention chirurgicale) ou de soins (dentaires par exemple). Le traitement de choix des malformations artérioveineuses pulmonaires dont le vaisseau afférent est ≥ 3 mm est l’embolisation (spirales métalliques) dans le même temps que l’angiographie, qui donne de bons résultats et supplante actuellement la chirurgie pulmonaire lourde et mutilante (3,4). ● Manifestations neurologiques La survenue d’un abcès cérébral doit faire rechercher systématiquement une MAV pulmonaire. Le traitement, rarement chirurgical, repose sur l’antibiothérapie(5). Le traitement des MAV fait appel à trois techniques utilisées seules ou en association : chirurgie, embolisation et stéréotaxie. Dans les formes asymptomatiques, une surveillance est discutée. ● Manifestations digestives et hépatiques En cas de syndrome hémorragique digestif, la coagulation des lésions télangiectasiques par laser sous endoscopie est le traitement de choix permettant de diminuer les besoins transfusionnels. La sclérothérapie peut permettre de juguler les hémorragies récidivantes nécessitant une supplémentation en fer. Des traitements estrogéniques ou progestatifs ont été proposés (6). La prise en charge des MAV hépatiques est complexe et souvent décevante. Le recours à l’embolisation ou à la chirurgie est actuellement exceptionnel. La transplantation hépatique est discutée en cas de cirrhose évoluée (7). Conclusion La morbidité de la maladie de Rendu-Osler est considérable et la mortalité est estimée à 10 % : elles sont principalement liées aux malformations artérioveineuses cérébrales et pulmonaires. Cette affection rare, d’expression variable avec le temps, nécessite une prise en charge multidisciplinaire. Des progrès importants ont été réalisés récemment dans le dépistage et la prise en charge thérapeutique de la maladie (mise en place d’études génétiques et techniques d’imagerie performantes peu invasives pouvant être plus facilement proposées aux malades), ainsi qu’une structuration des soins avec établissement de centres de compétences (liste disponible sur http://www.amro-france.org/).
Attention, pour des raisons réglementaires ce site est réservé aux professionnels de santé.
pour voir la suite, inscrivez-vous gratuitement.
Si vous êtes déjà inscrit,
connectez vous :
Si vous n'êtes pas encore inscrit au site,
inscrivez-vous gratuitement :